Géometrie et Quasi-cristaux¶
Le texte et les figures de cet article ont été inspirés/copiés d'après des articles de D. Gratias et le livre "Quasicrystal:A primer" par C. Janot (Oxford University Press)
Introduction¶
En 1982, Dan Shetchman découvrait les quasi-cristaux. L'alliage qu'il avait observé était parfaitement ordonné à longue distance mais présentait un ordre d'orientation (celui de l'icosaèdre) incompatible avec la nature périodique d'un cristal. Pour expliquer ce résultat surprenant, les physiciens d'alors ont du repenser et étendre les concepts de cristallographie. C'est ces quelques concepts fondamentaux que j'aimerais présenter ici, d'abord d'un point de vue purement géométrique puis ensuite appliqués à la structure quasi-cristalline réelle.
Les structures incommensurables¶
On pourrait penser que les quasi-cristaux sont les seules structures a être non-périodiques et ordonnées. En réalité nous allons commencer notre périple en s'intéressant aux structures dites "incommensurables" qui partagent des points communs avec les quasi-cristaux. Ceci constituera notre point de départ. Les structures incommensurables furent découvertes en 1936, mais prédites déjà en 1929. Elles détournent en quelque sorte le principe de périodicité à leur compte. Il faut s'imaginer une structure dans laquelle deux sous-structures de périodicité différente se superposent mais de telle sorte que le rapport dans leur période soit dans un rapport incommensurable, vous savez comme le rapport hauteur/largeur d'un rectangle d'or !
La figure ci-contre montre une structure "commensurable" et une "incommensurable" à 1 dimension. Si on superpose deux séquences de périodes commensurables (en haut le rapport est ½) et bien on obtient une structure qui est toujours périodique, seul le motif de base varie. Par contre si on superpose deux périodes incommensurables (en bas le rapport est racine carrée de 2) et bien aucune périodicité ne peut se retrouver dans la structure de base. Cependant ce type de structure ne fait que cacher sa périodicité !

Elle entretient un lien sous-jacent avec les cristaux que l'on ne rencontrera plus dans les quasi-cristaux, et c'est à ce titre qu'ils forment véritablement une catégorie à part. Cependant, la description des structures incommensurables fait appel à des concepts proches de ceux des quasi-cristaux, comme nous allons le voir.
En réalité, on voit bien que pour décrire la position des billes sur notre tige, il va nous falloir deux coordonnées. Une multiple de la période des billes noires et une multiple de la période des billes bleues, puisque les deux périodes sont incommensurables. Par exemple, si on prend l'origine (0,0) sur la bille noire de gauche, 1ère bille bleue aura comme coordonnée (0,1) et ainsi de suite... Ainsi il est plus commode de représenter les coordonnées dans un espace à 2 dimensions. On voit ici l'idée qui va sous-tendre la construction de ces structures: la périodicité se retrouve si on décrit la structure dans un espace de dimension supérieure.
Une autre façon de décrire la structure incommensurable serait de dire qu'il existe une seule période mais que celle-ci est modulée par une fonction périodique mais de période incommensurable avec la période de la structure initiale. La figure ci-contre nous présente une telle construction. Si la période de la fonction (ici sinusoïdale) est commensurable avec la structure initiale on obtient une structure également périodique en ajoutant des billes à l'endroit où la fonction coupe la tige. Dans le cas, de la figure du bas, le rapport est incommensurable. Ici, on pointe déjà un problème épineux. Dans une vraie structure les atomes ne peuvent pas se superposer comme dans le cas ici. Ceci implique des contraintes supplémentaires. On en reparlera plus tard. Il est facile de remarquer que si on déplace l'origine de la fonction (on parle de sa phase) et bien on retrouve une structure incommensurable que l'on ne peut distinguer de la précédente (si l'on considère la séquence infinie).

Ainsi, on peut regrouper toutes les structures d'une même "classe" ensemble. Comment pourrait-on s'y prendre ? Et bien comme nous l'avons vu on peut se servir de la notion d'espace à 2 dimensions. Pour générer une séquence à 1 dimension, il est juste nécessaire de couper notre espace à 2 dimensions par une droite appelée espace parallèle (R_{par}). L'ensemble des structures peuvent être empilées le long d'un axe perpendiculaire à l'axe de la chaîne. L'espace perpendiculaire ainsi décrit représente la variation de la phase de la fonction de modulation et peut être ainsi appelé espace de phasons.


Il est maintenant compréhensible que toute structure incommensurable peut être construite à partir de la coupe d'un réseau de dimension supérieure. De manière plus générale on va pouvoir construire une structure de dimension n à partir d'un espace de dimension n+1. On voit sur la figure ci dessus qu'il s'agit de couper astucieusement le réseau carré d'origine par l'espace physique. Cette représentation nous sera très utile dans la suite.
Dans le monde des solides, les cristaux se taillent une belle part, et exhibent une organisation rigoureuse basée entre autre sur le principe de symétrie de translation. Des structures non-périodiques peuvent être envisagées en détournant le principe de périodicité: ce sont les structures incommensurables qui peuvent se décrire par la superposition de deux réseaux périodiques mais de périodes incommensurables entre-elle. Ceci revient en fait à introduire une modulation dans la structure de départ de période incommensurable. Des structures semblables peuvent ainsi être obtenues en modifiant uniquement l'origine de la modulation (phase). Toutes ces structures semblables peuvent de façon plus générale être représentées dans un espace de dimension supérieure.
Fibonacci, Penrose, Ammann, Katz, Duneau et les autres¶
Dans cette section, nous allons parler de la manière dont on peut construire du moins de façon formelle des structures non périodiques. On s'intéressera en particulier aux concepts qui sont à l'origine de la description des quasi-cristaux. Pour plus de clarté on se placera soit à 1D soit à 2D. Il existe des exemples célèbres de structures non périodiques : la séquence de Fibonacci et le pavage de Penrose. Nous allons voir à travers ces deux exemples la manière plus générale de construire ce que l'on appelle des pavages apériodiques du plan.
La séquence de Fibonacci est une séquence alternant des segments courts (S) et des segments longs (L) construite en partant d’un segment donné et en posant la substitution suivante: L donne LS et S donne L. Si on part avec un segment long voilà ce qui se passe après plusieurs substitutions.


Si on souhaite maintenant obtenir un pavage non périodique du plan en utilisant le même principe de construction, la situation se complique singulièrement. On sait qu’il est impossible de paver le plan de façon régulière avec des pavés pentagonaux. Cependant en 1974, R. Penrose a réussi ce tour de force en réalisant un pavage non-périodique dans lequel la symétrie d'ordre 5 était globalement conservée (on reviendra sur ce point plus tard). C'est le fameux pavage de Penrose. Il est constitué de deux pavés élémentaires qui s'accolent entre eux par des règles précises qui assurent la bonne construction.

On conviendra de la difficulté de construire un tel pavage directement. On peut adopter un autre point de vue et construire des pavages par la méthode des multi-grilles (cf D. Gratias, Magazine Tangente (2002), 85 , 34–36 ). Une N-grille est un ensemble de N familles de droites sécantes. Chaque droite d'une famille intersecte une droite d'une autre famille en un point et un seul (grille régulière). La figure ci-contre présente une 3-grille.

La méthode des grilles permet de construire n'importe quel type de pavages du plan. La méthode en est très simple et se prête très bien au calcul par ordinateur. Elle associe à chaque grille un réseau dit dual (le pavage). Expliquons d'avantage comment on obtient ce réseau dual. On prend une N-grille et on collecte l'ensemble des vecteurs qui sont perpendiculaires à une famille de lignes. Cela forme un polygone. On joint les sommets au centre, ce qui nous découpe le polygone en secteurs. A l'intérieur de ces secteurs, on choisit un vecteur quelconque. On construit alors les parallélogrammes formés par les vecteurs pris deux à deux, associés à une famille de droites. Ainsi, on peut construire le pavage correspondant, qui n'est pas périodique ! Pas besoin de symétries 5 pour avoir des structures apériodiques ! On montre comme nous allons le voir que ces pavages sont le résultat de la coupe irrationnelle d'un objet de dimension N. Cette méthode des grilles permet de construire n'importe quel pavage. La figure ci-dessous vous présente deux exemples de pavages pentagonaux et octogonaux.



Lorsque les droites se croisent près d'un point triple, plusieurs situations peuvent être constatées comme cela est illustré sur la figure ci-contre. On remarque que la position des droites dans deux configurations très proches correspond à un renversement des configurations formées par les losanges. Ceci correspond à un "phason", imperfection dans l'arrangement des configurations. On reviendra sur ce point plus tard.
On vient de voir comment étendre cette méthode au pavage de symétrie 5 si on considère une 5-grille. On peut aussi prendre une grille dont les lignes sont espacées de façon quasi périodique, c'est à dire si l'espacement des lignes suit la séquence de Fibonacci ! C'est la grille d'Ammann. Elle est montrée sur la figure ci-contre. On y dénombre un nombre fini de cellules élémentaires. Elle permet de s'affranchir des cellules trop petites. Cette question est bien entendu importante dans les quasi-cristaux dans lesquels les distances entre atomes ne sont pas arbitrairement petites. Devinez quel est le réseau dual de la grille d'Ammann ? Le pavage de Penrose ! En formant le réseau, on y retrouve les deux types de losanges élémentaires.


Comme, nous l'avons vu dans la section précédente, il est intéressant de penser les structures non périodiques comme périodiques mais dans un espace de dimension supérieure. Cette idée est au cœur de la méthode utilisée pour construire les quasi-cristaux. C'est la "méthode de coupe et projection" proposé par Katz, Duneau et Elser principalement. Nous allons en donner deux exemples à 1D et à 2D.
Considérons un réseau carré périodique. La méthode de coupe et projection consiste à définir un espace de coupure que l'on appellera comme précédemment espace physique ou espace parallèle E_\| et l'espace complémentaire, espace perpendiculaire E_\perp. L'espace de coupe, ici une droite, n'est pas choisi au hasard. Dans ce cas la pente de la courbe vaut le nombre d'or ! Maintenant on enlève la partie inférieure du réseau et on projète les nœuds du réseau les plus proches sur la droite. On obtient la fameuse séquence de Fibonacci !


On peut procéder de la même façon en décorant le réseau de segments attaché à chaque nœud. La taille de ces segments est calculée de telle sorte que le déplacement de la droite le long de l'espace perpendiculaire, ne donne pas d'autres segments que ceux de la séquence de Fibonacci. On remarque à cet effet qu'une telle translation n'altère pas le caractère apériodique de la séquence. Les deux séquences sont indiscernables (on reviendra sur ce point par la suite).
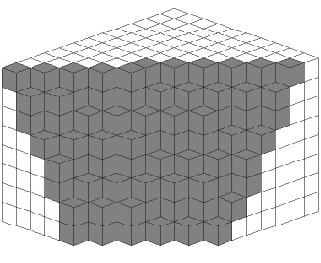
De même, si on se place dans un espace à 3 dimensions cubique, on peut réaliser la coupe et projection du réseau. La figure ci-contre en est un exemple. On peut la voir comme un réseau de cubes empilés formant des marches, soit comme un pavage du plan non périodique avec deux types de losanges. On voit ici l'équivalence entre méthode des grilles et celle de coupe et projection: le pavage est obtenu ici à partir d'une 3-grille (3 losanges différents) qui est aussi la coupe irrationnelle d'un espace de dimension 3 ! Dans le cas du pavage de Penrose on peut l'obtenir de la même façon à ceci près que l'espace périodique est à 5 dimensions ! Lorsque l'on considère un quasi-cristal icosaèdral comme nous le verrons plus tard, il nous faut un réseau périodique à 6 dimensions !
Nous avons vu ici comment construire d'une façon formelle des structures non périodiques, soit par une méthode directe, soit par la méthode dite des grilles ou soit par la méthode de coupe et projection. Cette dernière est celle que l'on emploie pour décrire la structure des quasi-cristaux. Elle est similaire à celle employée dans les structures incommensurables mais fondamentalement différente dans l'utilisation de "surfaces atomiques" discontinues.
La notion de symétrie dans les quasi-cristaux¶
Cette section est une adaptation d'un article de D. Gratias
Jusqu'à présent, nous nous sommes contentés de décrire les propriétés purement géométriques des pavages (et a fortiori des quasi-cristaux), mais que peut on dire de la notion de symétrie. Est-ce qu'un pavage de Penrose a une symétrie 5 ? Faisons le point sur les propriétés des pavages non-périodiques. La première propriété fondamentale est celle d'uniformité. Lorsqu'on observe le pavage de Penrose par exemple, on peut se convaincre que toute partie finie du pavage (aussi grande que l'on veut) se répète quasi-périodiquement une infinité de fois. La distance moyenne de répétition dépend de la partie choisie et croit avec sa taille, contrairement aux cristaux où cette distance est constante et égale à la période du réseau. En conséquence, le nombre de configurations différentes dans un cercle de rayon r est fini: tous les environnements locaux sont classifiables autour d'un point d'un pavé. C'est le principe d'uniformité.

La deuxième propriété remarquable est liée au déplacement de la droite de coupe le long de E_\perp comme nous l'avons déjà vu. Les structures obtenues sont similaires les unes par rapport aux autres; on dit qu'elles sont isomorphes. Le "théorème d'isomorphisme local" dit que toute partie finie de l'un des pavages se retrouvent dans l'autre. Si on combine les deux théorèmes, on a que les deux pavages sont essentiellement indiscernables sauf à l'infini ! Nous allons voir que ces deux théorèmes (uniformité et isomorphisme local) permettent de définir la notion de symétrie dans un pavage non-périodique. Pour comprendre observons la notion de symétrie de translation. Dans un cristal, une translation du réseau entraîne la superposition du réseau avec lui même: c'est la propriété d'invariance. Si on considère une translation d'une séquence 1D non-périodique, seule une fraction de points se recouvrent. Si on compte le nombre de points qui se recouvrent (n) dans un intervalle donné, le taux de recouvrement n/N (N, nombre total de points) tend vers une limite non nulle: un nombre infini de points se superposent.
Si, on raisonne à partir de la méthode de coupe et projection, on voit que la translation t est en fait la composante horizontale du vecteur \vec{T}, translation du réseau 2D. La séquence translatée peut être donc aussi obtenue à partir de la translation de la droite de coupe parallèlement à E_\perp. Les deux séquences sont localement isomorphes. Les points communs des deux séquences sont donc engendrés par l'intersection des mêmes segments verticaux. Pour connaître le taux de recouvrement, il suffit de calculer le taux de recouvrement entre deux segments translatés horizontalement l'un sur l'autre (rectangle gris). Comme la coupe est irrationnelle, le taux de recouvrement est compris entre 0 et 1 exclus. Si on fait l'algorithme de coupe et projection avec le petit segment d'intersection on obtient la séquence d'intersection qui est quasi-périodique. Le recouvrement partiel s'étend régulièrement jusqu'à l'infini.

Peut-on dire alors que la translation t est une opération de symétrie ? En réalité, puisque cette opération consiste en un déplacement dans l'espace perpendiculaire, des points sautent d'un site à un site voisin lorsque la droite de coupe passe par le centre d'un carré (ce saut élémentaire dont on reparlera est un phason). Cette opération revient à une inversion par rapport aux centres de carrés et qui se répète une infinité de fois ! L'opération de quasi-symétrie est donc définie comme celle qui transforme une séquence en une séquence localement isomorphe en laissant une fraction non nulle de points invariants. De la même façon, l'opération de rotation conduit à un nombre fini de points invariants. Sur la figure de gauche, un pavage de Penrose a subi une rotation de 72°. Les zones bicolores ne sont pas en coïncidence.
Ainsi, à la notion de symétrie classique de superposition parfaite du réseau, on est passé à une notion d'invariance locale: autour d'un point du pavage, l'environnement (atomique dans les quasi-cristaux) est indiscernable après une opération de symétrie, car quelque soit la taille de la zone que l'on inspecte, elle se retrouve une infinité de fois dans le pavage (uniformité et isomorphisme local)! Ainsi, le pavage de Penrose admet bien la symétrie pentagonale dans le sens où les propriétés physiques sont les mêmes dans toutes les directions équivalentes du pentagone. Dans les cristaux, les propriétés physiques dépendent de la symétrie au sens géométrique. Cette équivalence disparaît pour les quasi-cristaux. Nous allons reparler du rapport entre symétrie physique et géométrique dans la prochaine section.
Symétrie et Propriétés physiques¶
Avant de poursuivre notre chemin vers la compréhension des quasi-cristaux, nous allons nous arrêter quelques instants sur le lien entre la symétrie et les propriétés physiques. Ces considérations permettent d'investiguer les structures réelles et non plus les pavages ! La notion de symétrie de translation nous permet de définir un réseau à partir de 2 vecteurs de base dans le plan. Les vecteurs de base définissent ce que l'on appelle la "maille élémentaire", c'est à dire l'entité fondamentale permettant de construire l'ensemble de la structure, en tenant compte du motif (les atomes). Deux conséquences majeures ressortent de cette propriété. La première, purement géométrique, implique une restriction dans la symétrie de rotation (qui ne peut être que d'ordre 1, 2, 3, 4 et 6). La seconde est physique: si l'on envoie un faisceau d'électron ou de rayons X, les atomes diffusent le rayonnement dans toutes les directions de sorte que les ondelettes émises s'annihilent par interférence, comme les creux et les bosses de deux vagues se compensent entre eux. Cependant, du fait de la symétrie du cristal, certaines ondelettes interagissent de façon constructive. Elles donnent naissance au phénomène de diffraction. Ainsi le faisceau incident se scinde dans ces conditions.

Le phénomène de diffraction a donc lieu dans des conditions bien particulières qui sont réalisées lorsque la différence de phase entre les rayons diffractés par des atomes différents se combine de façon constructive. Pour cela, il est nécessaire que le déphasage entre les deux ondes soient un multiple entier de fois 2\pi. Or deux rayons diffractés par des atomes voisins ne vont pas parcourir la même distance jusqu'à l'observateur. La condition de diffraction est simplement réalisée lorsque le produit (\vec{k}'-\vec{k}).\vec{r}= \vec{q}.\vec{r} est entier. Cela correspond à la différence de trajet des deux rayons. \vec{q} représente en quelque sorte la quantité de mouvement transférée lors du "choc" comme deux boules se heurtant au billard.
On voit que les conditions de diffraction vont être réalisées que dans des directions particulières. L'interprétation de ce phénomène a beaucoup excité les physiciens au début du 20ème et a montré toute sa capacité à investiguer la matière. Quelle est la signification de ces conditions ?
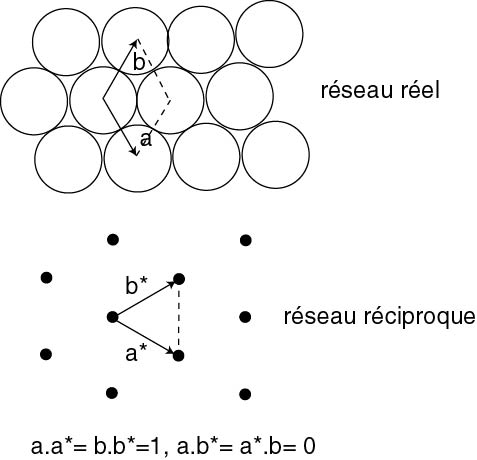
Pour déterminer les conditions de diffraction, nous allons être amenés à faire le même type d'opération que l'on a rencontrée dans la méthode des grilles: on va construire un réseau dual du réseau réel. La figure ci dessous montre cette opération pour un réseau hexagonal. En haut, on a représenté le réseau dit réel, comprenant les deux vecteurs de base \vec{a} et \vec{b} qui définissent la maille élémentaire (en pointillé). Cette maille est décorée d'atomes (ronds). La transformation dual est ici un peu différente: les vecteurs de base sont modifiés de telle sorte que \vec{a}^\star et \vec{b}^\star soit respectivement perpendiculaire à \vec{b} et \vec{a}, et que la longueur des vecteurs \vec{a}^\star et \vec{b}^\star soit l'inverse de celle des vecteurs \vec{a} et \vec{a}. On peut tracer le réseau réciproque (en bas). On voit clairement qu'il a les mêmes symétries que le réseau réel (en particulier on reconnaît la symétrie hexagonale). Revenons à notre diffraction. On sait que \vec{r} est un vecteur du réseau réel, par conséquent l'équation \vec{q}.\vec{r}=n implique que le vecteur \vec{q} est un vecteur du réseau réciproque !
Ainsi, lorsque l'on va "éclairer" un cristal avec un rayonnement, on pourra obtenir ce que l'on appelle un cliché de diffraction (ensemble de tâches) qui sera donc la cartographie du réseau réciproque et donc caractéristique de la nature du réseau cristallin (il faut prendre en compte également le motif atomique).
Les cristaux sont construits à partir d'une maille élémentaire, décorée d'atomes, que l'on reproduit dans toutes les directions du plan ou plus généralement de l'espace par l'opération de translation. Cette opération de symétrie, implique des restrictions d'ordre géométriques mais plus surtout définit les conditions de diffraction. On montre que les faisceaux diffractés se concentrent sur les nœuds d'un réseau qui a les mêmes symétries que le réseau réel. Ceci permet donc un parfaite cartographie du réseau.
Diffraction et Quasi-cristaux¶
Jusqu'à présent, nous avons essentiellement parlé de pavages et de leurs propriétés géométriques. Nous allons maintenant nous intéresser aux quasi-cristaux et en particulier les phases icosaèdrales. La notion de diffraction va nous aider à mieux appréhender ces phases.
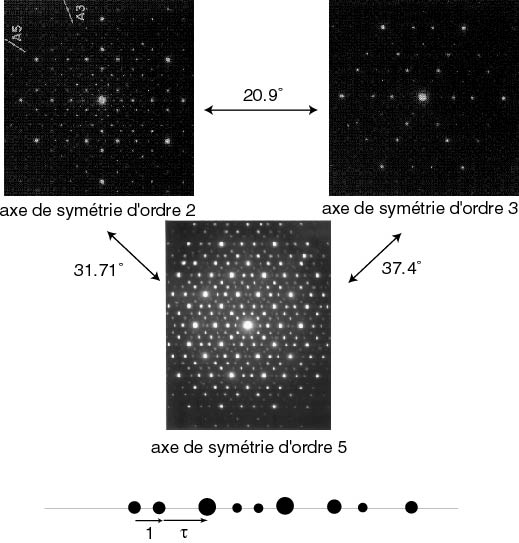
Lorsque l'on observe une structure icosaèdrale telle que l'alliage Al(70%)Pd(20%)Mn(10%), on observe un cliché de diffraction essentiellement ponctuel, preuve que la structure est ordonnée à longue distance. Elle présente essentiellement trois figures de diffraction exhibant les symétries de l'icosaèdre (analogue 3D du pentagone), à savoir, des axes de symétrie binaire, ternaire et quinaire (5). On dit que le "groupe ponctuel" est celui de l'icosaèdre. Si on regarde de plus près on pourra se convaincre que dans une direction, les intensités des tâches de diffraction sont différentes et se répartissent de façon quasi-périodique.
Pour comprendre comment indexer un diagramme de diffraction quasi-cristallin, repartons de l'exemple à 2D. Considérons par exemple que le réseau réciproque soit décrit par un pentagone. On voit clairement, que pour définir tous les nœuds du réseau il va falloir 4 vecteurs de bases, deux dans chaque direction et qui sont incommensurables entre eux (leur rapport qui est le nombre d'or provient des propriétés même du pentagone). On comprend ainsi qu'un quasi-cristal 2D exhibant une symétrie pentagonal (par exemple le pavage de Penrose) peut être construit à partir d'un espace à 4 dimensions !

En effet si on calcule le diagramme de diffraction d'un pavage de Penrose on retrouve un cliché caractéristique de symétrie 5. De plus, on voit ici que, du fait de l'irrationalité des vecteurs de base, on va pouvoir construire des vecteurs aussi petits que l'on veut. Ceci signifie simplement que, comme chaque vecteur correspond à une condition de diffraction, le spectre de diffraction va être dense et non plus ponctuel !
Il est pourtant clair que le spectre de diffraction n'a pas l'air dense et comporte un nombre fini de tâches de diffraction. Essayons de voir pourquoi. Si on prend un espace périodique à 2D pour effectuer la méthode de coupe et projection, il est nécessaire de décorer les nœuds du réseau par ce que l'on a appelé des "surfaces atomiques" (comme on décore les cristaux par des atomes). On reparlera plus tard de ces surfaces, mais il est clair qu'elles sont allongées parallèlement à l'espace perp. Jusqu'à présent lorsque l'on a décrit la diffraction, on a pris en compte que la nature géométrique du réseau et non la présence du motif atomique qui lui peut être différent pour un même réseau. Ceci a pour conséquence d'introduire une contribution supplémentaire à l'intensité diffractée, et permet donc de différencier des structures. Dans notre cas, le terme introduit par nos surfaces atomiques est un terme décroissant très rapidement et qui tend vers 0 à l'infini. En fait, si l'objet est ponctuel, l'intensité diffractée se concentre aux nœuds du réseau réciproque, mais si la surface est allongée l'intensité diffractée est étalée mais se concentre principalement au voisinage des nœuds du réseau. Si on représente le réseau réciproque de l'espace périodique carré (qui lui aussi est carré !), on peut voir l'intensité diffractée dans l'espace physique comme la coupe irrationnelle de ce réseau en tenant compte de la présence des surfaces atomiques comme sur la figure ci-contre.
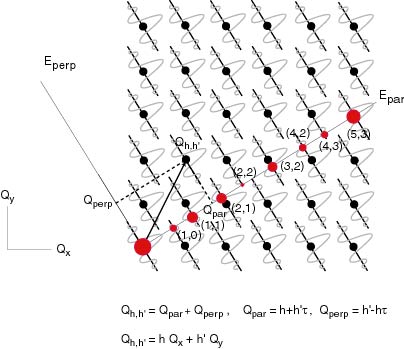
On voit bien que le spectre est dense ce qui est du au fait que le terme provenant des surfaces atomiques décroît sans s'annuler totalement. Cependant, seules quelques tâches sont réellement visibles. On voit également ici que les tâches de diffraction s'indexent avec 2 vecteurs dans l'espace parallèle et dont le rapport des longueurs est le nombre d'or (résultat de la coupe irrationnelle d'un espace de dimension 2). Ainsi 2 indices sont nécessaires pour décrire la position de chaque tâches dans l'espace physique (du au fait de l'irrationalité des longueurs), ces 2 indices étant les coordonnées des nœuds dans l'espace carré. Le même raisonnement peut être appliqué dans le cas de quasi-cristaux icosaèdraux: il faut 6 vecteurs diffraction pointant vers les sommets d'un icosaèdre pour décrire l'espace réciproque. Ainsi la structure de ces quasi-cristaux est obtenue par coupe et projection d'un espace "hyper-cubique" à 6 dimensions ! Ainsi tous les vecteurs seront définis par 6 entiers (n_1,.....,n_6) dans cette maille. Dans l'espace physique il faut 6 indices également, s'écrivant de la forme (h+h'\tau, k+k'\tau, l+l'\tau), 2 indices par direction étant nécessaires.

La diffraction du rayonnement par une structure quasi-cristalline diffère radicalement de la diffraction par une structure cristalline du fait même que l'on ne puisse définir un réseau réciproque périodique. Celui-ci peut se définir par un ensemble de 6 vecteurs pointant vers les sommets d'un icosaèdre définissant la classe des quasi-cristaux icosaèdraux. Ainsi cette structure peut être vue comme la coupe à 3D d'un espace de dimension 6 ! Dans l'espace physique, les coordonnées des vecteurs diffraction sont définies par 2 indices dans chaque direction pour tenir compte de l'irrationalité des vecteurs entre eux.
Mais où sont les atomes ?¶
Cette section est tirée de et du livre "Quasi-crystals: A primer" (chapitre 3 et 4) par C. Janot
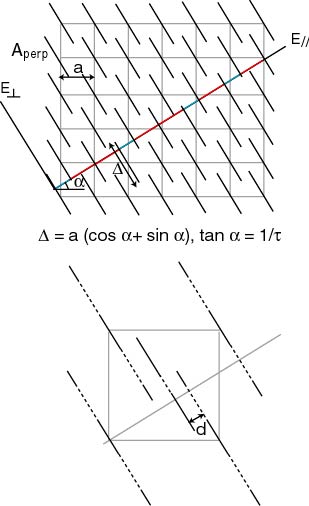
La question que l'on est en droit de se poser maintenant est de savoir où se trouvent réellement les atomes dans la structure ? Cette question est plutôt délicate. La difficulté réside dans le fait qu'il faille se placer dans un espace de grande dimension dans lequel un grand nombre de paramètres doivent être définis. Dans une première étape, on s'attache à définir les surfaces atomiques A_{\perp} dans l'espace périodique. Dans ce but, quelques remarques doivent être observées.
Si on se place dans un espace à 2D, on voit clairement que:
-
les surfaces atomiques doivent être "plates" dans la direction de E_\perp pour générer dans l'espace physique des atomes ponctuels.
-
elles ne peuvent pas intersectées l'espace physique en s'approchant les unes des autres à une distance minimum (reliée à la taille des atomes).
-
elles doivent être invariantes lorsqu'on déplace l'espace de coupe dans E_\perp. Ceci signifie que les surfaces doivent se connecter de telle sorte qu'il n'y ait aucune création ni d'annihilation d'atomes lors de la translation. Cette condition restreint la taille de la surface A_\perp dans E_\perp.
-
à quoi on peut rajouter qu'elles sont invariantes par opérations de symétrie de la structure. Cependant, le nombre et la position de ces surfaces doivent être également déterminés. Pour se fixer les idées, considérons non plus un réseau carré décoré de segment à chaque nœud mais un réseau décoré en plus par un segment au centre de chaque carré. La façon dont on peut éviter les distances inter-atomiques trop petites (d) est de créer un trou dans la surface du centre et un anneau dans les surfaces aux nœuds. Cette solution n'est pas unique mais a le mérite de définir la façon dont on construit les surfaces atomiques.
Pour connaître la structure dans l'espace physique, il est facile de déterminer les positions atomiques par l'algorithme de coupe et projection. Cependant, cette description n'est pas pertinente. D'après ce que l'on a déjà vu, dans une boule de rayon r, il existe un nombre de configurations données et qui se répètent une infinité de fois et comme la structure est localement isomorphe, ces configurations se retrouvent dans la structure après une opération de symétrie. En dressant ce que l'on appelle un "r-atlas", il est plus pertinent de définir la structure comme l'ensemble des configurations les plus fréquentes. Cette opération peut être effectuée en faisant la "décomposition en cellules". Un exemple en est donné dans le cas de la séquence de Fibonacci.

Lorsque l'on définit la coupe et projection dans un réseau carré on peut définir des cellules L et S comme les domaines d'intersection entre les surfaces atomiques. On aboutit alors à définir les deux distances caractéristiques L et S de la suite de Fibonacci. Il est intéressant de voir que chaque L est entouré d'au moins un S et que chaque S à un 2 L pour voisins. Ainsi, toute la structure peut être définie par un enchevêtrement de clusters LS ou son symétrique SL. Ainsi toute l'information nécessaire à la description d'un quasi-cristal est définit dans ces cellules élémentaires. Par exemple dans le cas 1D, l'enchaînement de la séquence LS... y est défini.
Pour l'instant nous avons vu que le cas du quasi-cristal 1D ! La tâche est moins évidente pour une structure 3D telle AlPdMn. Le but est de trouver un ensemble de surfaces atomiques plates dont la forme et la localisation dans l'espace à 6D de manière à obtenir une densité, une composition et un spectre de diffraction qui correspondent aux données expérimentales. On va voir que cela restreint les choix possibles.
Un des problèmes majeurs dans la compréhension des quasi-cristaux est de savoir comment l'ordre à longue distance peut se propager. Quelles règles contraignent cet ordre ? Si on fait la comparaison avec les pavages non-périodiques comme celui de Penrose, on est amené à penser que l'ordre se propage de façon locale par accolement des tuiles dans un ordre bien défini. Cependant il existe des situations où deux tuiles différentes peuvent être choisies, une seule conduisant à une structure non-périodique. Ce choix ne peut se faire que par l'inspection globale du pavage, ce qui est difficilement concevable si l'on suppose que les interactions dominantes sont à courte portée. Toutefois on sait que des règles locales existent. Il a été démontré que l'on peut construire un quasi-cristal respectant un ensemble de règles locales par un choix de surfaces atomiques limités par des plans de symétrie miroir. Pourtant les lois locales ne sont pas des lois de croissance. Lors de la croissance, de nombreux défauts vont être générés, et le coût énergétique associé à la réparation de ces défauts (sous forme de phasons) doit être le plus faible possible. C'est la condition déjà évoqué précédemment: c'est celle d'invariance par translation dans l'espace perp. ! Ces deux conditions sont satisfaites pour des surfaces atomiques bordées par des plans miroirs. Si on prend en compte que la symétrie des surfaces atomiques est celle de l'icosaèdre, cela réduit le choix !
On peut maintenant se poser la question de savoir comment déterminer complètement ses surfaces. Ce point est un peu technique et ne sera pas discuté en détail ici. Les méthodes classiques de cristallographie échouent dans le cas des quasi-cristaux. On fait alors appel à des méthodes de diffraction X (analyse de Patterson) et de neutrons. Voyons brièvement de quoi il s'agit. La fonction de Patterson résulte en quelque sorte de la superposition de la carte de densité atomique (on parle de fonction d'auto-corrélation) et est obtenue à partir de l'intensité diffractée. Supposons dans notre cas que l'on ait deux surfaces atomiques aux nœuds du réseau (L_1) et un au centre (L_2).

On voit que la fonction de Patterson a dans ce cas une extension dans E_\perp . On comprend qu'il y a deux corrélations possibles: une sur les nœuds et une au centre (\rho_1 et \rho_2). Pour un déplacement dans E_\perp nul la superposition est maximale. \rho_1 décroît continûment jusqu'à 2L_2 valeur pour laquelle les segments L_2 ne se superposent plus. En conséquence \rho_1 diminue encore jusqu'à 2L_1.Pour connaître la corrélation du centre, on translate le réseau selon la diagonale en amenant les deux surfaces en coïncidence. Pour un certain déplacement il y a une intersection entre les deux segments et la fonction reste alors constante. Quand le déplacement atteint la valeur L_1-L_2, le recouvrement décroît jusqu'à L_1+L_2. Ainsi la fonction de Patterson est ici en parfaite correspondance avec la forme des surfaces atomiques. Elle a deux contributions situées aux nœuds du réseau et en son centre.
La correspondance n'est pas toujours aussi simple, mais permet de localiser les surfaces atomiques. La figure suivante montre la fonction de Patterson pour un quasi-cristal icosaèdral (AlPdMn): elle représente ici une coupe à 2D de l'espace périodique selon un plan de symétrie 5. On remarque la présence des surfaces atomiques allongées en forme de cigares selon E_\perp.
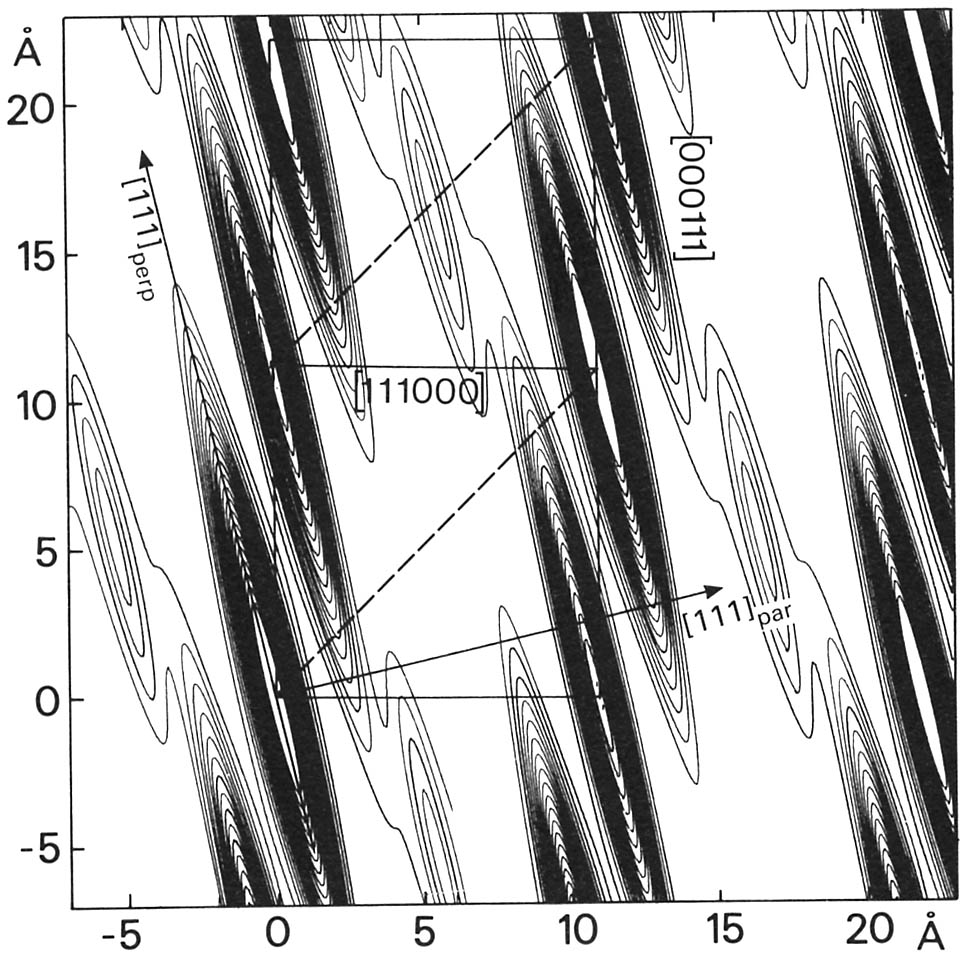
Une analyse minutieuse permet de dresser la répartition des composés en effectuant des analyses de diffraction de rayons X et de neutrons sur des composés dans lesquels on a substitué des éléments par d'autres. Ceci a pour effet de faire varier les contrastes de diffraction. On établit ainsi les cartes de Patterson pour les différents composés. Un modèle simple consiste alors à assimiler en première approximation, les surfaces atomiques à des sphères. La répartition des éléments peut être ainsi déterminée de façon à correspondre aux données expérimentales de diffraction.

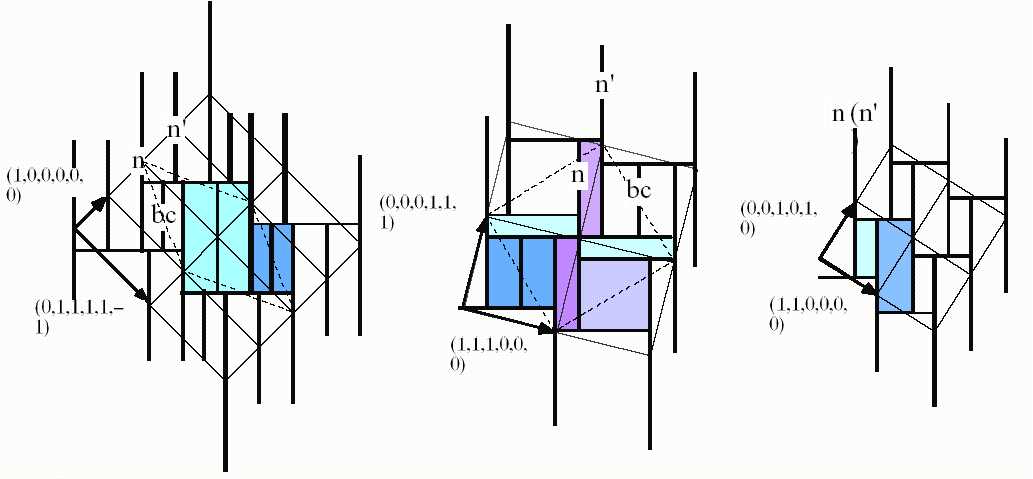
Une fois ce travail effectué, il s'agit de facetter les surfaces atomiques pour éliminer les distances inter atomiques trop petites en tenant compte des conditions géométriques précédemment établies. Ainsi, on trouve les 3 polyèdres représentés ci-contre, respectivement n, n' et bc.


Dans l'espace 6D, les trois surfaces atomiques sont attachées aux nœuds du réseau cubique (n et n') et au centre de certaines mailles dites primitives (bc). Ceci est illustré dans le cas d'une coupe dans un plan de symétrie 5.
On dit ici que le réseau est cubique primitif (maille élémentaire) possédant une super-structure (la nature des surfaces est différente au nœud n' et en plus certains centres de maille bc sont occupés). On dit dans ce cas que la structure est ordonnée (du point de vue chimique)
Reste à savoir comment se répartissent les atomes dans l'espace réel ! Bien sûr, il est possible partir de l'algorithme de coupe et projection pour générer l'ensemble des coordonnées des atomes, mais cette approche n'est pas la plus pertinente. Comme nous venons de voir il est plus approprié d'effectuer la "décomposition en cellules". La figure de droite représente la coupe à 2D de la structure selon des plans de symétrie respectivement de gauche à droite 5, 3 et 2. Les surfaces atomiques intersectent chaque plan selon des lignes qui s'étendent selon E_\perp. Les distances inter atomiques sont les projections parallèles entre deux nœuds qui ont une intersection non nulle dans E_\perp. On voit par exemple ici que les trois distances les plus courtes sont: n-bc (plan 5, cellule bleue foncée), n-n' (plan 2, cellule violette) et n-n (plan 2, cellule bleue claire). Toutes ces distances forment des configurations entre voisins que l'on doit analyser pour en dégager les plus fréquentes.
Ce travail révèle que 95% des atomes sont regroupés dans deux types de clusters comprenant 33 et 50 atomes. Ils s'interpénètrent de façon à formée une structure dont la symétrie moyenne est celle de l'icosaèdre. La compacité de ces structures est proche de celle rencontrée dans la plupart des alliages métalliques (qui sont eux des cristaux).
Les quasi-cristaux icosaèdraux tel AlPdMn, exhibent dans leurs clichés de diffraction, la symétrie de l'icosaèdre. Il est alors aisé de comprendre qu'ils doivent être décrits dans un espace à 6 dimensions du moment où 6 vecteurs indépendants doivent être choisis pour décrire entièrement l'espace réciproque. Déterminer la structure atomique exacte est une tâche difficile. Le choix des surfaces atomiques est guidé par des arguments relatifs à l'ordre local de la structure et de la minimisation de l'énergie des phasons, défauts qui sont associés au processus de croissance. L'analyse en diffraction X et de neutrons donnent des surfaces atomiques allongées selon l'espace perpendiculaire en accord avec le fait que l'intersection de ces surfaces avec l'espace physique donnent des atomes ponctuels. En rapprochant les quasi-cristaux des pavages non-périodiques en disant que dans une boule de rayon r, il n'existe qu'un nombre donné de configurations atomiques locales, on peut donner une représentation dans l'espace réel de la structure en décrivant les quelques amas atomiques les plus fréquents.
Croissance, quasi-cristaux et approximants¶
Les quasi-cristaux tels ceux qu'avaient observés à l'origine Dany Shechtman n'étaient pas de très bonne qualité. Depuis les méthodes d'élaboration ont progressé et on compte un grand nombre de composés différents principalement des alliages ternaires mais également quelques alliages binaires. Il existe de nombreuses techniques de préparation mais sont relativement délicates en raison de la relative instabilité des phases quasi-cristalline par rapport à des composés cristallins de composition proche.
Pour élaborer un cristal de façon plus générale, on part de ses composants dans des proportions définies sous forme d'un mélange liquide à très haute température. Dans la "méthode de Czochralsky", on plonge une tige dans le mélange qui sert de germe à la croissance du cristal que l'on retire du bain en fusion lentement. La différence de température étant faible, le mélange cristallise en une phase unique le long de l'axe du germe. On parle de croissance dirigée. Dans le cas de certains quasi-cristaux comme AlPdMn, on obtient des mono-quasi-cristaux de taille centimétrique. Un recuit à haute température est cependant nécessaire pour éliminer certaines phases cristallines.

Cliché B.Grushko, IFF, Jülich

Certains quasi-cristaux comme AlCuFe ne sont pas suffisamment stables à haute température pour être solidifier par la méthode de Czochralsky. On procède alors de la façon suivante: on envoie le mélange en fusion sur une roue froide en rotation rapide. Le taux de refroidissement est énorme, environ 1 million de degrés par seconde ! On obtient alors un ruban ou plus généralement une poudre quasi cristalline qui est ensuite compactée (frittage) pour faire coalescer les grains entre eux sous l'effet de la pression et de la température. On obtient alors des poly-quasi-cristaux dont la taille des grains est de l'ordre du micromètre. D'autres techniques permettent la croissance de quasi-cristaux exhibant leur symétrie d'orientation (symétrie macroscopique), comme celle du dodécaèdre ! C'est par exemple la méthode de la géode que l'on rencontre naturellement dans le cas du quartz.
Lorsque l'on regarde les quasi-cristaux à l'échelle atomique en microscopie à haute résolution, on se rend compte que les colonnes d'atomes forment des figures non-périodiques. Des pavages non-périodiques reproduisent dans ce cas très bien le motif observé dans un plan donné.

Cliché Université de Marburg
L'image suivante montre la superposition entre une image prise en microscopie à champ proche et les positions des atomes données par le modèle. L'accord est plutôt satisfaisant !

Cliché Luc Barbier, CEA
Jusque là on a parlé uniquement de quasi-cristaux icosaèdraux. Il existe des phases de type octogonal (8), dodécagonal (12) et décagonal (10) pour lesquelles le caractère quasi-cristallin est confiné dans 2 directions de l'espace, la troisième étant périodique ! C'est le cas par exemple de Al(70)Ni(10)Co(20) qui est une phase décagonale. Son cliché de diffraction est quasi-périodique et de symétrie 10 et périodique selon une direction.


Cependant, les phases quasi-cristallines sont loin d'être parfaitement stables. En réalité, elles vieillissent inexorablement ... car elles sont en fait métastables à température ambiante et tendent vers un état d'équilibre qui n'est pas quasi-cristallin en un temps fini mais qui est heureusement extrêmement long ! Cela dit, il existe des phases relativement proches des quasi-cristaux, mais qui sont périodiques. Ce sont des cristaux dont la maille élémentaire est très grande. On les appelle des phases approximantes. Pour comprendre de quoi il s'agit, imaginons que dans notre exemple de coupe et projection d'un quasi-cristal 1D, on modifie la droite de coupe de sorte que sa pente soit rationnelle.

Si on désigne par (n,m) les coordonnées du vecteur directeur de la droite, on peut former des approximants se rapprochant de plus en plus du quasi-cristal si on prend (n,m) comme les termes successifs de la suite de Fibonacci. On parle d'approximant 1/1 ou 3/2....On voit en tout cas que plus le rapport n/m tend vers le nombre d'or, plus la période (et donc la maille) est grande. On peut constater que la structure d'un approximant peut être obtenue à partir de la structure du quasi-cristal en appliquant un cisaillement linéaire le long de l'espace perp.
Lorsque l'on parle de quasi-cristaux, ce terme englobe une foule de composés classables essentiellement en composés quasi-cristallins dans les 3 directions de l'espace (les quasi-cristaux icosaèdraux) et ceux qui sont formés d'un empilement périodique de plans quasi-cristallins de symétrie 8, 10 et 12. Leurs propriétés thermodynamiques en font des composés difficiles à préparer. Toutefois, il n'est pas rare de synthétiser des mono grains de plusieurs centimètres dont la structure est parfaitement quasi-cristalline. D'autres composés répondant au nom de phases approximantes sont comme leur nom l'indique des structures périodiques de grande maille de structure proche à celle d'un quasi-cristal mais de nature différente.
Dislocations et Quasi-cristaux¶
NB : la lecture de ce paragraphe nécessite la connaissance de la notion de dislocation dans les cristaux. Se reporter à l’article : L'énigme de la Plasticité des Métaux avant la lecture de ce paragraphe
Pour finir intéressons nous à la notion de dislocation dans les quasi-cristaux. Cette notion y est clairement transposable si on considère la structure dans l'espace périodique à ...6 dimensions bien sûr. L'introduction d'une dislocation dans un milieu périodique engendre une déformation localisée autour du cœur de la dislocation. Que se passe-t-il dans le cas des quasi-cristaux ?
Reprenons notre analogie du début de quasi-cristal à une dimension. Il y a deux façons de déformer localement cette structure: en introduisant un déplacement local le long de l'espace physique (d_\|) soit le long de l'espace perpendiculaire (d_\perp). Dans le premier cas, il est clair que localement, certains segments seront raccourcis ou allongés. Ceci est tout à fait analogue à l'introduction d'un champ élastique (ou champ de "phonons"). Par contre, dans le second cas, localement certains segments n'occupent plus la bonne place, il y a des inversions (ou "flip") entre les pavés élémentaires. Ceci correspond aux phasons que nous avons déjà rencontrés ! Ils représentent en quelque sorte une perturbation d'ordre chimique dans la structure.

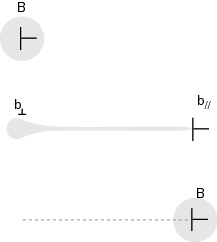
Le champ de déformation associé à un déplacement non uniforme dans l'espace perpendiculaire est appelé ainsi "champ de phasons". On montre ainsi que lors l'introduction d'une dislocation dans un quasi-cristal deux champs de déformation sont créés. Le premier est dit propagatif, c'est un champ d'origine élastique qui se propage sous la forme de vibrations du réseau, tandis que le second est dit diffusif car il implique le mouvement collectif d'atomes dans la structure. Une dislocation sera donc caractérisée par un vecteur de Burgers dans l'espace à 6D (\vec{B}), se décomposant en une composante élastique (\vec{b_\|}) et une composante de phasons ($\vec{b}_\perp $).
L'introduction d'une dislocation peut se faire naturellement si l'on considère le processus de Volterra. Toutefois, il est possible dans ce cas de l'effectuer de deux façons différentes. La première consiste à effectuer l'opération dans l'espace physique. Ceci est représenté sur la figure ci-contre dans notre analogie de quasi-cristal à deux dimensions. On obtient ici une dislocation coin en enlevant un demi quasi- plan et en recollant les lèvres de coupure d'une quantité bpar. Les quasi plans (en noirs) sont analogues aux plans cristallographiques classiques. Ils sont en réalité obtenus par l'intersection des plans cristallographiques dans l'espace périodique par l'espace physique ce qui leurs confère un caractère rugueux, notez aussi qu'ils ne sont pas équirépartis mais leur distribution suit la séquence de Fibonacci.


La dislocation obtenue est entourée d'un champ élastique du à la distorsion des quasi-plans autour du cœur. Cependant, dans le sillage de la dislocation, un certain nombre de mauvais accolements (mais qui préserve tout de même la "connectivité" du pavage) sont produits. En effet, ici dans une portion du pavage, on a représenté les règles d'accolements des pavés entre eux (flèches rouges). On peut voir ainsi que dans la zone de recollement, les pavés collés ne présentent pas les mêmes règles entre eux (flèches opposées). Ces mauvais accolements peuvent être vus comme une condensation de phasons élémentaires dans une faute. On parle ainsi de faute de phasons derrière une dislocation imparfaite (ou partielle) décrite par un vecteur de Burgers bpar qui n'est pas une translation du réseau à 6D. On la représente symboliquement comme sur la figure ci-dessus.
Toutefois, on peut également très bien effectuer le processus de Volterra dans l'espace périodique puis projeter la structure obtenue dans l'espace physique. Ceci est représenté ci-contre dans le cas d'un quasi-cristal à 2D obtenu par la coupe et projection d'un réseau cubique. Un demi-plan supplémentaire en rouge est cette fois inséré pour obtenir une dislocation coin de vecteur de Burgers \vec{B}. La structure obtenue dans l'espace physique est représenté ci-contre. On note toujours la présence d'un champ de déformation élastique autour du cœur (pavés élémentaires déformés), mais on remarque la présence de phasons individuels repartis autour de la dislocation.

Pour visualiser ce qu'est un phason ici, il est facile par comparaison avec la structure parfaite de remarquer qu'il résulte de l'addition ou la suppression d'un cube élémentaires, ou si l'on préfère de l'inversion des configurations hexagonales formées par les pavés élémentaires ("flip"). On obtient ainsi une dislocation parfaite (de vecteur \vec{B}) représenté comme ci-contre.

Si l'on regarde une telle dislocation à très haute résolution, on peut voir apparaître les phasons entourant le cœur de la dislocation. Ceux ci se présentent sous la forme de "jags" traduisant les inversions de configuration de même nature que ceux dont nous avons parlé précédemment. Rappelez-vous le formalisme de la grille pour décrire un pavage non-périodique (par exemple le pavage de Penrose). Vous souvenez vous du fait qu'une inversion de configurations (c'est à dire des phasons) provenaient d'un déplacement d'une ligne.
On voit facilement qu'une famille de lignes constitue une famille de "vers" (analogues aux plans) fortement rugueux formés dans le cas du Penrose par un ensemble de configurations hexagonales. Lorsque l'on introduit un phason, on effectue une inversion dans l'arrangement des vers, ce qui revient à un décalage d'une ligne de la grille à l'endroit même du flip. En haute résolution, ou l'on voit les plans atomiques moyens de tels décalages ou "jags" sont clairement visibles.
La figure ci dessus montre: en haut, un phason comme le résultat d'un flip d'une configuration de pavés accompagné d'une erreur d'accolement et le "jag" associé dans le formalisme multigrille. Au milieu, la représentation symbolique d'une dislocation parfaite dans un pavage de Penrose. Les grilles permettent de se rendre compte du champ élastique (lignes courbés) et du champ de phason ("jags") autour du cœur de la dislocation (espace non pavé). En bas, vue rasante d'une dislocation vue en haute résolution. Les "jags" autour de la dislocation apparaissent comme une discontinuité des plans atomiques moyens (points blancs).
Depuis les années 90, les recherches se sont intensifiées sur les propriétés mécaniques des quasi-cristaux grâce à l'étude des dislocations dans ces matériaux. Dans AlPdMn, il est maintenant fermement établi que la plasticité est assurée par la multiplication et le mouvement des dislocations. Que ce passe t'il exactement quand on applique une contrainte (mécanique ou thermique) dans un tel environnement ? Comment se déplacent les dislocations ?
Et bien, les dislocations interagissent avec cette contrainte par l'intermédiaire de leur composante de champ élastique (\vec{b}_\|). Ci-dessous est représenté le mouvement d'une telle dislocation. On part d'une configuration où la dislocation est parfaite entourée de son champ de phason (en gris). L'application de la contrainte entraîne la décomposition de la dislocation en une dislocation imparfaite traînant une faute de phasons dans son sillage et laissant la composante de type phason immobile (\vec{b_\perp}). Si la température est assez élevée, la faute tend à s'étendre en dehors de la faute et si la dislocation vient à s'arrêter, le repavage total de la faute s'effectue conduisant à la situation d'origine.
La figure ci-dessous à droite montre deux dislocations imparfaites traînant une faute de phason. Dans leur sillage une longue dislocation est également visible. Celle ci apparaît comme un ruban composé de franges brillantes et sombres. Remarquer en particulier que les franges bordant la faute sont noires (indiquées par les flèches).

Pour connaître les paramètres caractéristiques de la plasticité du matériau, il est nécessaire de connaître le mode de déplacement des dislocations. Celui-ci est défini par le plan de déplacement des dislocations et leur vecteur de Burgers.
Dans le cas bien connu du déplacement par glissement, le vecteur de Burgers est un vecteur du plan de déplacement : le mouvement se fait par la propagation d’un cisaillement sans déplacement de matière. Lorsque le vecteur de Burgers est hors du plan, on a affaire à un déplacement par montée. Ce type de mouvement correspond à la dissolution ou l’accroissement du demi- plan supplémentaire ajouté pour former une dislocation coin. Il s’effectue donc grâce à la diffusion d’atomes sur de longues distances. Comme en général, la diffusion des atomes est facilitée par l’agitation thermique, la montée des dislocations est rencontrée dans les cristaux qu’à haute température.

Dans les quasi-cristaux, on a d'abord pensé, qu’à l'instar des cristaux, le mécanisme dominant était du glissement. Toutefois, des études menées au CEMES, ont montré que le mécanisme de montée était responsable de la plasticité des quasi-cristaux, même à basse température, et que le glissement pourrait être interdit dans ce genre de structures. Cette propriété pourrait en particulier expliquer le caractère extrême fragile des quasi-cristaux à température ambiante comme la conséquence de l'impossibilité de mouvement de dislocations.
Les preuves expérimentales de montée peuvent être de plus étayées par des considérations purement géométriques. Si l'on raisonne dans un pavage apériodique du plan, tel le pavage de Penrose, il est facile de voir qu'un cisaillement le long d'un "quasi-plan" va créer de nombreuses fautes qui ne pourront être repavés. Dans le cas de la montée, on peut procéder par la duplication ou la disparition de ces quasi-plans. Dans ce cas bien qu'une faute soit créée (due aux mauvais accolements), la topologie du pavage est préservée !

Il existe encore de nombreux enjeux dans la compréhension des phénomènes fondamentaux qui régissent le comportement de tels matériaux et ils sont loin d'être tous démasqués. A travers l'étude des dislocations et des propriétés mécaniques, nous avons découvert finalement que la particularité structurale de ce matériau permettait le renouvellement des conceptions fondamentales attachées au monde traditionnel des cristaux et en retour, de redécouvrir des mécanismes d'apparence marginale (la montée) mais qui jouent indéniablement un rôle considérable à haute température dans les alliages métalliques. Mais on ne pouvait attendre mieux des quasi-cristaux tant ils font preuve d'originalité !